Síndrome de Stevens-Johnson
Síndrome de Stevens-Johnson
Publicado em 29 de abril de 2015
Historicamente, o eritema multiforme, a síndrome de Stevens-Johnson (SSJ) e a necrólise epidérmica tóxica (NET) faziam parte de um mesmo espectro clínico. Mas, recentemente, há uma tendência para se considerar os eritemas multiformes minor e major como partes de um espectro, frequentemente relacionados à infecções (especialmente herpesvirus) e ocasionalmente devido à reações a drogas. Já a SSJ e a NET estão mais ligadas à reações a drogas (80-95%) e são consideradas variantes severas de uma mesma doença.
A incidência da SSJ é de 1 a 6 e a da NET de 0,4 a 1,2 casos por milhão de pessoas ao ano. Nos dois quadros os padrões imunohistológicos das lesões iniciais sugerem uma reação citotóxica mediada por células contra as células epidérmicas, levando à apoptose.
Quando o destacamento epidérmico envolve < 10% da superfície corporal (SC) os pacientes são classificados como SSJ, > 30% SC como NET e entre 10 e 30% SC como formas transicionais. Há relatos de maior frequência de SSJ e NET em pacientes com AIDS.
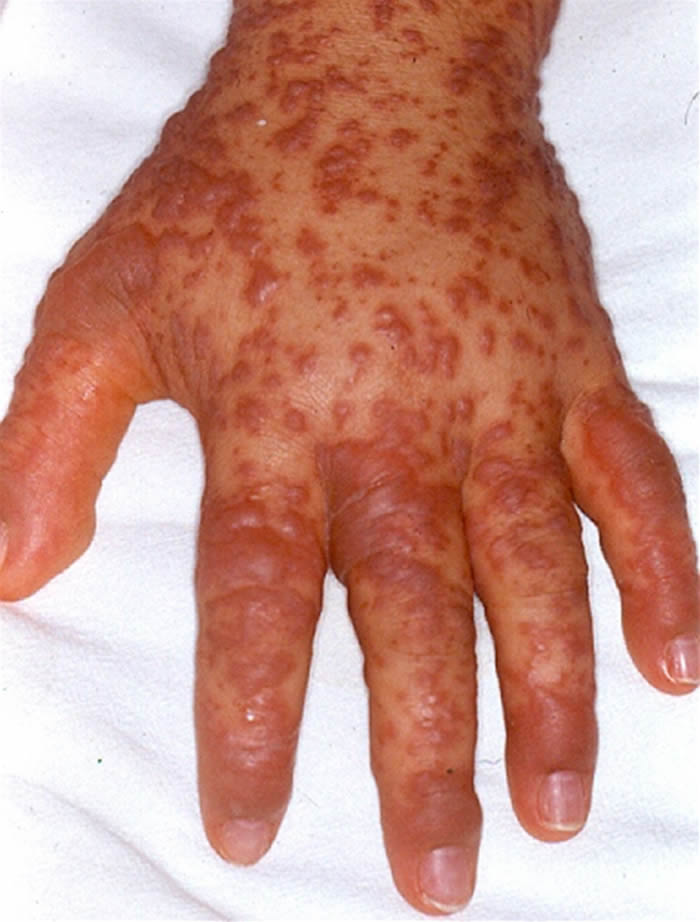
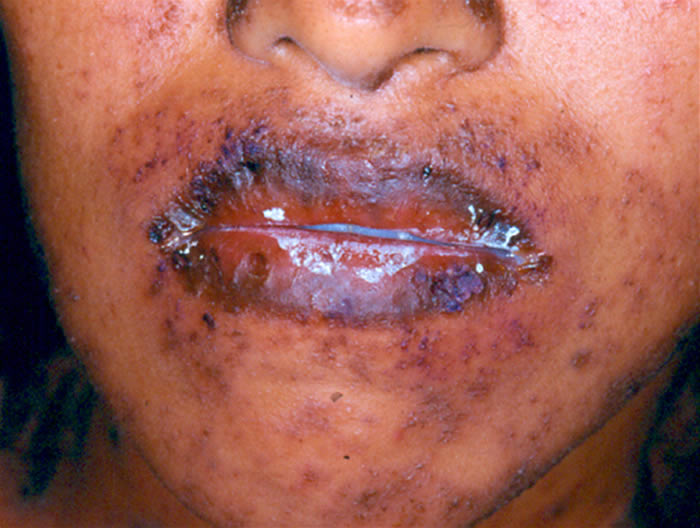
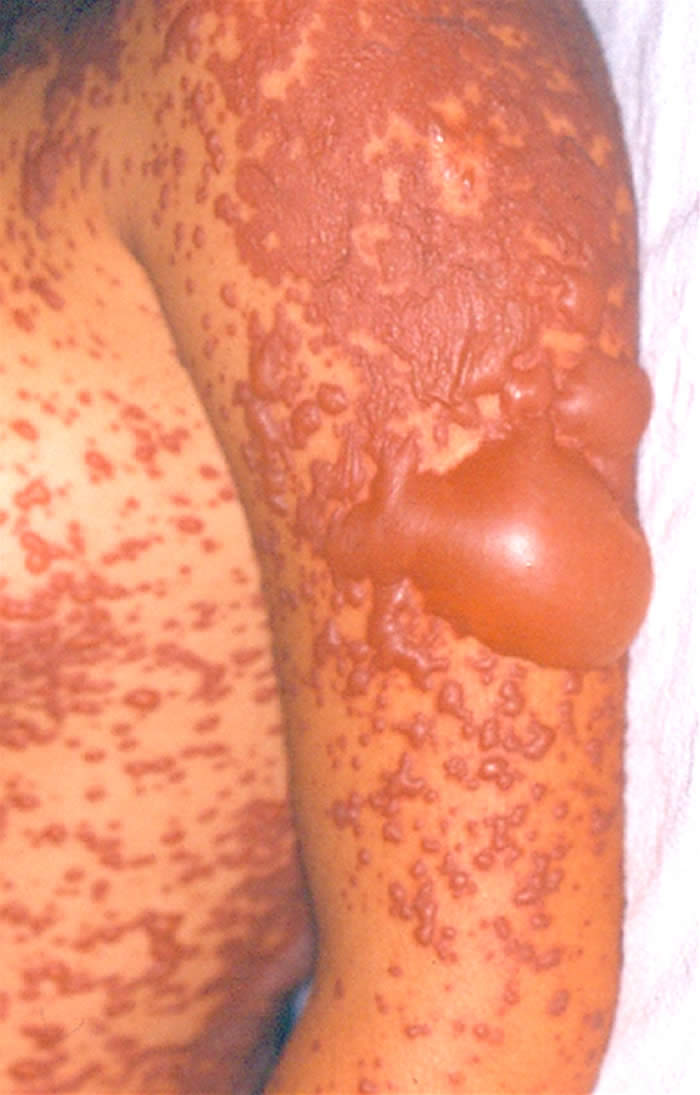
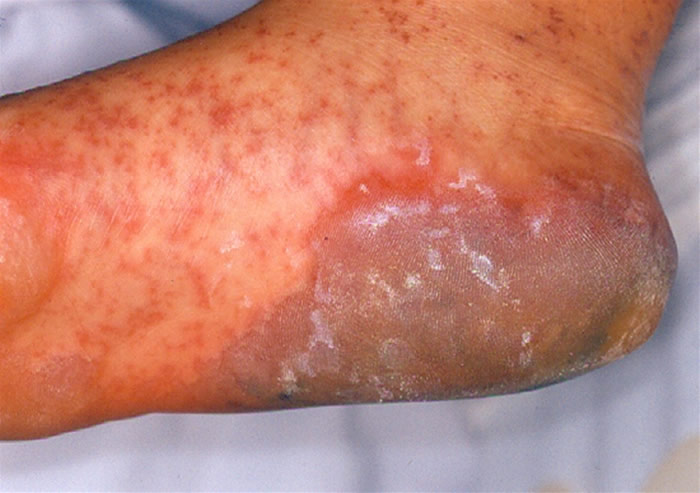
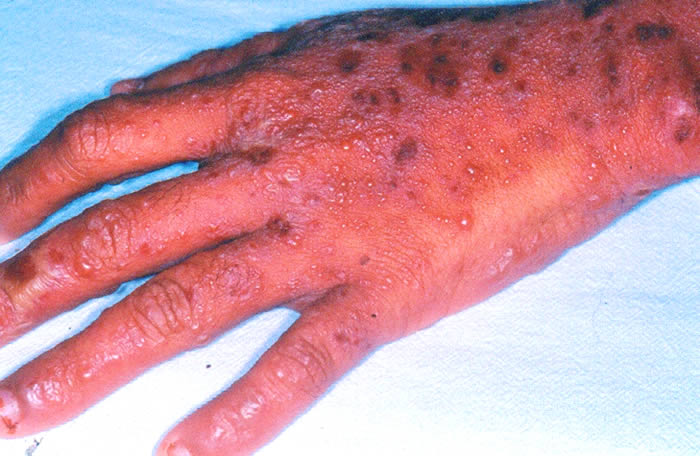
Síndrome de Stevens-Johnson: Foi descrita em 1922. O quadro tem início geralmente de forma súbita, porém sintomas prodrômicos (1-14 dias) podem surgir antes do aparecimento da erupção cutânea, que é variável em extensão. São vistas lesões características de extensivo eritema multiforme, principalmente no tronco, do tipo máculo-papuloso, urticariforme, bolhoso e até mesmo pustuloso. As membranas mucosas são acometidas, normalmente 2-3: oral 100%, ocular 90%, genitália masculina 57% e anal 5%. A mucosa oral apresenta extensa formação de bolhas que erosam e se recobrem por membrana branco-acinzentada e crostas hemorrágicas.
O comprometimento ocular é freqüente podendo ocorrer severa conjuntivite purulenta, ulceração da córnea, uveíte anterior e panoftalmia. Pode ocorrer acometimento visceral, com broncopneumonites, traqueítes, hemorragias gastrointestinais e glomerulonefrite com necrose tubular aguda. Embora haja centenas de etiologias, as drogas são os principais agentes causais, especialmente os antiinflamatórios não-hormonais (ibuprofen e naproxen), sulfas, anti-convulsivantes (hidantoínas e barbitúricos) e antibióticos. Estas drogas são metabolizadas no fígado e na pele através de um sistema similar, incluindo o sistema de citocromos p450, gerando óxidos arênicos, que em indivíduos susceptíveis, ligam-se ao RNAm e inibem a síntese protéica celular. Na SSJ associada à droga, as lesões mucosas se iniciam de 14-56 dias após a ingestão do medicamento.
Assim, as drogas ingeridas poucos dias antes do início do quadro, usualmente não são implicadas. O diagnóstico diferencial com a doença de Kawasaki nas crianças e com o pênfigo paraneoplásico nos adultos deve ser lembrado. No tratamento, fora as medidas gerais, o uso de corticóides sistêmicos e da imunoglobulina intravenosa ainda é debatido.